
Utilizing the first-in-class STEP delivery technology for in vivo genome editing, CourageAS develops groundbreaking therapeutics to transform the lives of individuals with Angelman syndrome.
Angelman Syndrome
Angelman syndrome (AS) is a rare genetic neurologic disorder caused by the loss of function in the UBE3A gene in the maternal chromosome 15q11-q13 region. In a neurotypical individual, the maternally inherited UBE3A gene is expressed, while the copy of the gene inherited from the father is silenced, only in neurons, by a phenomenon known as genomic imprinting. For individuals living with AS, alterations in the maternal UBE3A gene result in a lack of or deficiency in expression. Genotypes include maternal gene deletion, genetic point mutation, uniparental disomy, imprinting center defect or somatic mosaicism.

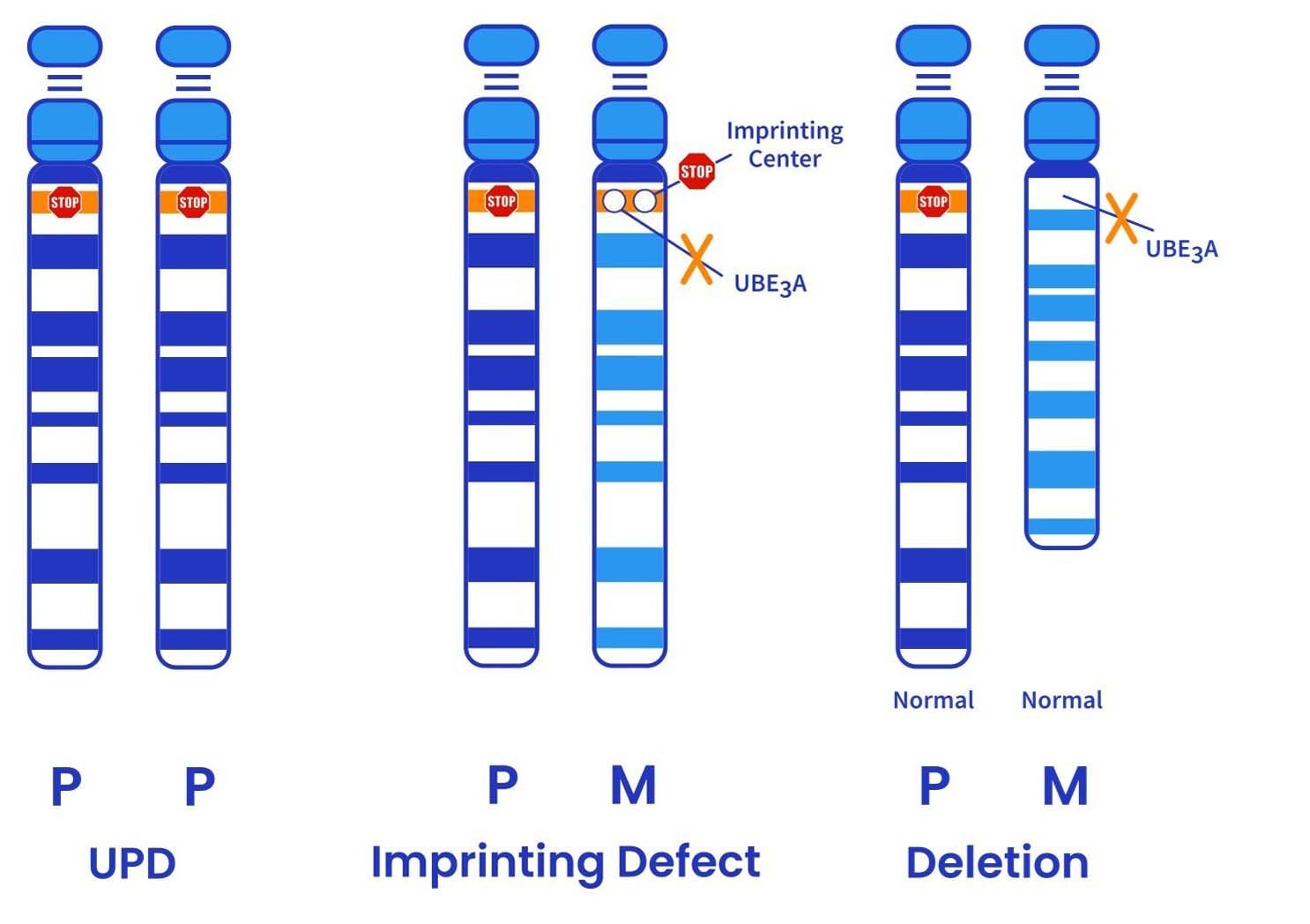
Source: FAST (cureangelman.org)
There are no approved disease-modifying treatments for AS. Reactivating the silenced paternal UBE3A gene holds great promise for potentially treating this devastating disorder. The expression of UBE3A in neurons can be activated through inhibition of the silencing mechanism, a long non-coding RNA known as UBE3A antisense transcript (UBE3A-ATS). Early-stage clinical trials with investigational antisense oligonucleotides (ASOs) targeting UBE3A-ATS, have demonstrated promising safety and efficacy results which further support a paternal activation strategy.

Direct delivery of the CRISPR/Cas9 genome editor into the CSF fluid via a ribonucleoprotein (RNP) complex could represent a promising approach to treatment of neurogenetic disorders. CourageAS is developing an investigational AS genome-editing candidate utilizing Couragene’s Stimuli-Responsive Traceless Engineering Platform (STEP) technology to deliver an enhanced CRISPR/Cas9 RNP complex to disrupt the UBE3A-ATS gene. This approach may achieve widespread brain delivery without the concern for chronic nuclease exposure when delivered via viral vector. Early preclinical data has shown that a single administration of this STEP-RNP results in brain-wide, persistent reactivation of UBE3A from the paternal chromosome in both an AS mouse model and human patient-derived iPSC cell lines. In addition, a single administration of STEP-RNP enables efficient expression of paternal Ube3a to ameliorate neurobehavioral deficits in a maternal Ube3a-deficient AS mouse model when dosed at all ages tested (newborn, juvenile, and adult). Encouraged by these promising preclinical data, we aspire to harness the power of transformative STEP-RNP technology to deliver a potential option for individuals living with AS.